May 28, 2026

The first six months of 2026 rebuilt the framework medical device manufacturers spent the last three years learning. QSIT gave way to QMSR. The FCC's IoT labeling program lost its anchor tenant. The EU Cyber Resilience Act moved from theory into compliance calendars. FDA issued its first publicly visible warning letter explicitly citing a cybersecurity vulnerability handling failure. FDA leadership turned over mid-stream. And AI inserted itself into both the products being regulated and the regulators reviewing them, while simultaneously lowering the barrier for attackers to find and exploit vulnerabilities.
This post breaks down what happened across three lenses (regulatory, financial, technical), then looks ahead to what the second half of 2026 will demand.
The single largest regulatory event was the Quality Management System Regulation (QMSR) effective date. FDA published a substantially rewritten Compliance Program Manual (CP 7382.850) to replace QSIT, with direct cybersecurity implications:
Documentation that was "good enough" under the old framework may not survive a read through the ISO 13485 and ISO 14971 lens. FDA's premarket cybersecurity guidance was also updated to align references, though substantive policy appears largely unchanged.
The FCC's IoT labeling program lost UL after the new administration opened an investigation into UL's China operations, leaving the Cyber Trust Mark program's future unclear.
The EU Cyber Resilience Act (CRA) compliance clock is also ticking loudly. MDR/IVDR-regulated devices are excluded, but the ecosystem around them generally is not. Companion apps, cloud dashboards, OTA servers, and most peripherals are in scope, and mandatory vulnerability reporting begins September 11, 2026.
Reports described "cracks showing" at CDRH. Industry pressed for FDA staffing transparency in MDUFA discussions, and Commissioner Marty Makary resigned in May. None of this helps predictable review timelines.
On the EU side, an EU-commissioned study found more than half of EU medical device companies have reduced product portfolios to cope with MDR/IVDR complexity and 17% have stopped producing devices entirely — the macroeconomic backdrop against which every additional EU obligation, CRA included, now lands.
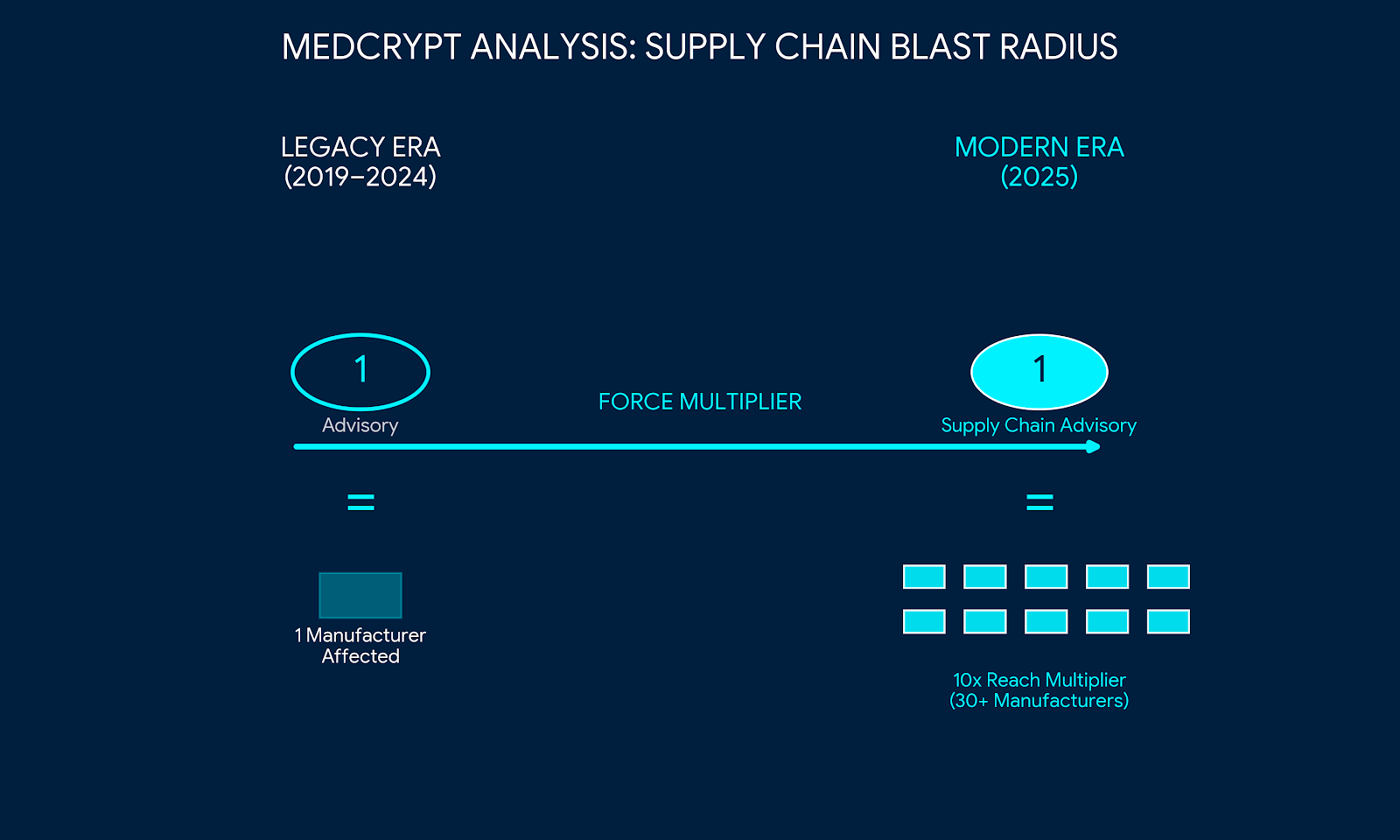
The Beta Bionics warning letter, published January 28, is, as far as we can tell, the first publicly visible FDA warning letter to explicitly cite a cybersecurity-related failure. The specifics:
The legal substrate matters. Failure to comply with cybersecurity requirements under Section 524B is a prohibited act under 21 U.S.C. §331(q)(3), enforceable through injunction, seizure, and criminal prosecution. The cost-of-compliance versus cost-of-non-compliance math just got more concrete.
Separately, FDA started asking for VEX and VDR files alongside CycloneDX SBOMs post-shutdown, pushing manufacturers toward continuously maintained vulnerability statements rather than point-in-time submissions.
Three trends defined the technical half.
MITRE published new resources on SBOM normalization and cybersecurity risk analysis for medical devices in the era of cloud, AI/ML, and PQC. The G7 Cybersecurity Working Group released a positioning paper on AI-BOM. CISA published guidance on OpenEoX for communicating software lifecycle stage, dovetailing with BOD 26-02 on end-of-support edge devices. Germany's BSI published TR-03185-2 on open-source software lifecycle management. The connecting thread: transparency about what is in a device, what state it is in, and wha
FDA recognized AAMI CR515 on cybersecurity for AI/ML-enabled medical devices. In a notable first, FDA issued a warning letter to Purolea Cosmetics Lab explicitly citing the use of AI to generate "product specifications, procedures, and master production or control records" without process validation.
The reverse case matters too: if AI tooling becomes standard for Notified Body review, expect 100% inspection of submitted documentation — every fault caught, not just sampled ones.
The UK NCSC published timelines requiring PQC migration for all systems, services, and products by 2035, with priority migration by 2031 and migration goals set by 2028. That is the first hard date many manufacturers will have seen on PQC.
If the first half of 2026 was about transitions taking effect, the second half is about their consequences converging.
Regulators and Notified Bodies will increasingly run AI-assisted reviews of technical documentation, and small inconsistencies previously missed will be flagged systematically. Invest in AI tooling of your own to pre-flight submissions, and treat consistency and traceability across the dossier as a first-class engineering concern. The Purolea warning letter is the preview.
The targeted HIPAA Security Rule update and the steady drumbeat of state privacy laws (Alabama, Oklahoma, and Kentucky all moved in early 2026) point the same direction. Cybersecurity controls need to extend beyond confidentiality and availability into demonstrable integrity. That means logging, signing, attestation, and auditability of both product data and submission data.
Several forces are converging on the postmarket side:
Buyer-side pressure is already showing up in the numbers. RunSafe's 2026 Medical Device Cybersecurity Index found that 84% of healthcare organizations now build cybersecurity requirements into procurement and 56% have rejected devices on security grounds, up from 46% a year ago. For manufacturers, that means the evidence package that satisfies FDA increasingly has to do double duty as a sales asset.
Capable general-purpose reasoning models (Anthropic's Mythos being the most-discussed recent example, with OpenAI and others moving the same direction) can now pinpoint and build working exploits for software vulnerabilities as a side effect of broader reasoning gains, not as a dedicated capability.
The practical implication, laid out by Oleg Yusim, is that LLM-based systems make vulnerability discovery easier and accessible to everybody, leading to rapid development of new zero-day exploits. Anthropic's own Project Glasswing update has since echoed the same warning.
The defensive math does not survive this unchanged:
For medical device manufacturers, that is not a tooling change. It is a re-architecting of the entire release pipeline, with testing, validation, regulatory documentation, and secure over-the-air delivery all on the critical path. Treat that work as a 2026–2027 program, not a 2028 one.
If they are not already there:
A recurring observation this half: "you're always responsible for the code, whether your cat wrote it, your LLM wrote it, or your team wrote it." That principle applies cleanly to the broader cybersecurity posture of a medical device organization in 2026.
The regulatory machinery is being rebuilt. The standards landscape is being consolidated. The enforcement tools are being tested in public. The technical demands are expanding into AI provenance and post-quantum readiness simultaneously. None of that changes the underlying obligation. All of it changes what discharging that obligation looks like in practice.
The work is the same. The scrutiny is not.

July 9, 2026


April 15, 2026
March 12, 2026
Get the latest healthcare cybersecurity news right in your inbox.
We'll never spam you or sell your information


